Le nouveau règlement européen des essais cliniques (Règlement (UE) n° 536/2014), entré en vigueur en janvier 2022, représente une avancée majeure pour la recherche biomédicale en Europe. Son objectif principal est de renforcer la transparence, l’efficacité et la sécurité des essais cliniques tout en simplifiant les procédures administratives pour les chercheurs et les entreprises pharmaceutiques. Ce règlement vise à harmoniser les processus d’autorisation des essais cliniques à travers les États membres, réduisant ainsi les délais et les coûts associés à la mise en place de nouveaux essais.
Une des nouveautés clés de ce règlement est la nouvelle approche d’analyse en deux parties du dossier clinique, qui concerne la soumission et l’évaluation des demandes d’essais. Le dossier est désormais divisé en deux sections distinctes :
- La partie scientifique, qui inclut les données de préclinique, les protocoles des essais, ainsi que les justifications scientifiques pour les méthodes et la conception de l’étude. Cette partie est évaluée par un comité d’experts, qui examine la validité scientifique et méthodologique de l’essai proposé.
- La partie éthique, qui se concentre sur la sécurité des participants, le respect des normes éthiques et la protection des données personnelles. Cette partie assure que l’essai respecte les normes éthiques en vigueur, en particulier en ce qui concerne le consentement éclairé des participants, ainsi que la gestion des risques liés à l’essai clinique.
Avantages et inconvénients du nouveau règlement européen

D’autre part, le « plan santé innovation 2030 » a pour objectifs :
- Simplification et accélération de l’autorisation des essais cliniques
- Valorisation de l’expertise de l’évaluation éthique des CPP
- Alléger la charge du CPP en spécialisant certains CPP au traitement des dossiers médicaments Europe
- Simplifier et clarifier le rôle des CPP et de l’ANSM
- Création d’un guichet commun par l’ANSM
- Augmenter les moyens des CPP et RiPH
- Harmonisation des pratiques des CPP
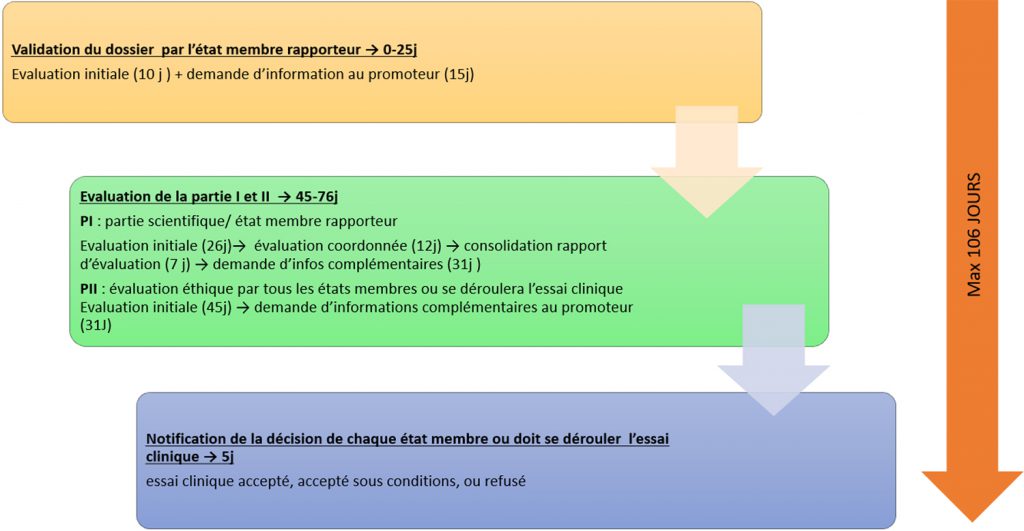
Après dépôt du dossier de l’essai clinique par le promoteur sur le portail européen CTIS, les délais d’instruction sont fixés pour chaque étape d’évaluation .
Délais des différentes étapes d’évaluation du dossier d’essai clinique déposé par l’industriel